Research
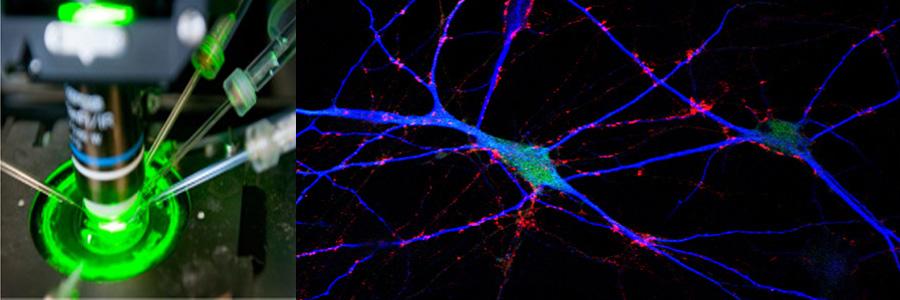
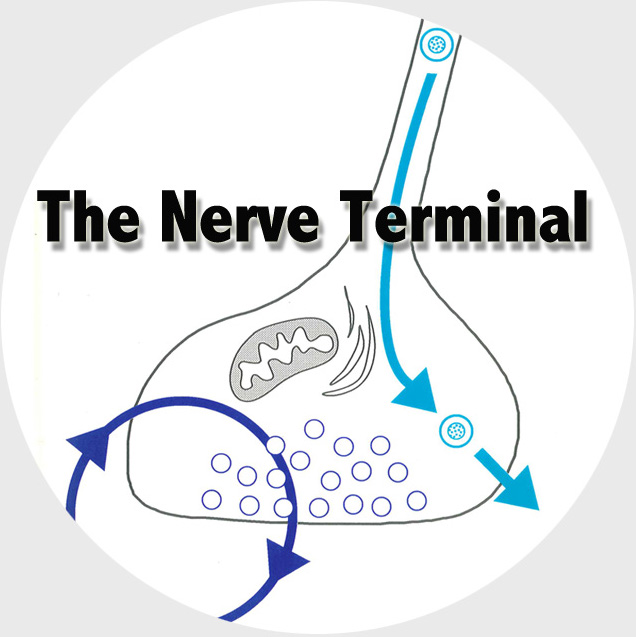
The Functional Genomics department studies the presynaptic nerve terminal in health and disease.
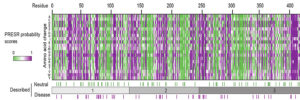
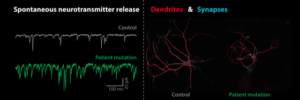
We study presynaptic mechanisms that adjust synaptic transmission (presynaptic plasticity), the trafficking & fusion of neuropeptide vesicles (dense core vesicles) and presynaptic mechanisms of degeneration.